Rev Cuid 2017; 8(1): 1391-406
doi: 10.15649/cuidarte.v8i1.373
EDITORIAL
APPLICATION OF MENDELIAN RANDOMIZATION: CAN WE ESTABLISH CAUSAL RISK FACTORS FOR TYPE 2 DIABETES IN LOW-TO-MIDDLE INCOME COUNTRIES?
APLICACIÓN DE LA ALEATORIZACIÓN MENDELIANA: ¿PODEMOS ESTABLECER FACTORES DE RIESGO CAUSALES PARA LA DIABETES TIPO 2 EN PAÍSES DE BAJO Y MEDIO INGRESO?
APLICAÇÃO DA RANDOMIZAÇÃO MENDELIANA: PODEMOS ESTABELECER FATORES DE RISCO CAUSAIS PARA A DIABETES TIPO 2 EM PAÍSES DE BAIXA E MÉDIA RENDA?
Ryan James Quentin Langdon1, Kaitlin Hazel Wade2
[Full Spanish and Portuguese text after the English text]
1BSc, Integrative Epidemiology Unit, University of Bristol, Bristol, United Kingdom. Corresponding author, E-mail: ryan.langdon@bristol.ac.uk
2PhD, Integrative Epidemiology Unit, University of Bristol, Bristol, United Kingdom, E-mail: kaitlin.wade@bristol.ac.uk
History
Receipt date: November 18, 2016
Approval date: December 16, 2016
How to cite this article: Langdon RJQ, Wade KH. Application of Mendelian randomization: can we establish causal risk factors for type 2 diabetes in low-to-middle income countries? Rev Cuid. 2017; 8(1): 1391-406.http://dx.doi.org/10.15649/cuidarte.v8i1.373
![]() © 2017 Universidad de Santander. This is an Open Access article distributed under the terms of the Creative Commons Attribution- NonCommercial (CC BY-NC 4.0). This license lets others distribute, remix, tweak, and build upon your work non- commercially, as long as they credit you for the original creation.
© 2017 Universidad de Santander. This is an Open Access article distributed under the terms of the Creative Commons Attribution- NonCommercial (CC BY-NC 4.0). This license lets others distribute, remix, tweak, and build upon your work non- commercially, as long as they credit you for the original creation.
The global burden of type 2 diabetes (T2D) is increasing, partially facilitated by a sharp increase in the disease in low and middle income countries (LMICs)1,2. LMICs not only show a high prevalence of T2D (8.7%), but have shown a much faster increase in this prevalence over the past 30 years when compared to high-income countries (HICs)1. Conventional risk factors for T2D in HICs, such as high body mass index (BMI)3, low levels of physical activity4, and poor dietary behaviours5, do not fully account for the greater increase in prevalence of T2D in LMICs2. Therefore, risk factors for T2D specifically within an LMIC context need to be determined.
Observational epidemiological analyses are currently being used to explain the rising prevalence and determine outstanding risk factors for T2D in LMICs, but even if designed well, such studies are prone to confounding, reverse causation and multiple sources of bias (e.g. selection, reporting and measurement)6. As such, these study designs can potentially generate unreliable estimates of causality between a risk factor and disease. A successful, more robust approach to overcome these limitations and improve causal inference is Mendelian randomization (MR)6,7.
Briefly, arising from instrumentable variable (IV) analyses in econometrics, MR exploits Mendel’s first and second laws of inheritance (i.e. the independent assortment and segregation of alleles that at leads to the random distribution of genotypes in the population) enabling the use of genetic variants to proxy for a clinically relevant (and usually modifiable) trait (e.g. BMI)6,7. Provided a number of key assumptions are met (Figure 1), these genetic IVs can then be used to derive the causal effect on a trait on disease or adverse health outcome, conferring multiple advantages over observationally-derived estimates of association.
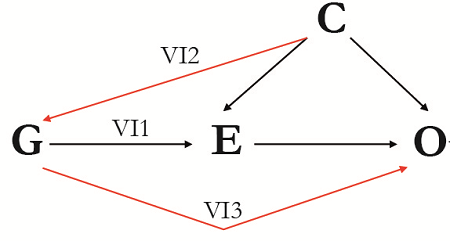
Figure 1. Directed acyclic graph (DAG) of the theory and key assumptions of Mendelian randomization. A genetic variant (or variants, G) can be used as instrumental variables for an exposure of interest (E) to assess the causal association between E and the outcome of interest (O) given that the following three assumptions hold: (IV1) G must be robustly associated with E; (IV2) G must not be associated with any measured or unmeasured confounding variable (C); and (IV3) there must be no independent association between G and O, given E and C.
Analogous to arms of a randomized control trial (RCT), genetic variants used in MR are largely independent of confounding factors due to the random nature of their allocation within a population. They are also not modified by the later development of disease or health outcome (Figure 2) and, with the advent of more accurate genotyping arrays, measurement error is largely reduced. Therefore, at a population level, the portion of variance in the modifiable trait explained by genetic variants used as an IV (unlike the direct measurement of the trait) is free of the limitations that would otherwise weaken causal inference in observational studies. MR provides a robust, unconfounded estimate of the casual association between a trait and disease6,7.
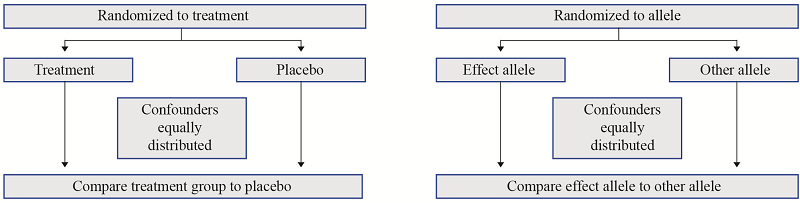
Figure 2. Mendelian randomization methodology compared to randomized controlled trials (RCTs). In an RCT, individuals are randomized to either the treatment or control (placebo) arms, which theoretically ensures that confounders are equally distributed among trial arms. Analogous to this, the random allocation and independent assortment of alleles at conception and meiosis, respectively ensure that confounders are equally distributed among genotype groups. Differences in individuals between genotype groups will differ only due to the genotype group; hence, we can use genetic variants as instrumental variables for exposures of interest in Mendelian randomization analyses.
In the context of HICs, MR has confirmed and identified endogenous risk factors for T2D including obesity, systemic inflammation and blood lipids, and exogenous risk factors including alcohol and dairy consumption8,9. For example, the use of MR in a recent comprehensive assessment of the causal association between BMI and T2D produced refined causal estimates suggesting that T2D risk increased by approximately 30% with each unit (kg/m2) in BMI (odds ratio (OR): 1.26; 95% confidence interval (CI): 1.19, 1.32; P=3.29x10-10)10. However, these findings cannot reliably be translated to LMICs. Until recently, the expense and availability of large samples of genetic and phenotypic data restricted MR analyses to HICs; a majority of which reside within populations of European ancestry. Therefore, whilst it is likely that higher adiposity may, at least in part, explain the growing prevalence of diabetes in LMICs, the estimate of association remains untested and currently only a few MR studies exist that assess the impact of any modifiable exposure on disease risk specifically in LMICs.
Specifically within the context of Latin America, Borges et al.11 investigated the causal effect of circulating homocysteine levels on blood pressure in the 1982 Pelotas Birth Cohort in Brazil using MR, then compared these results to those from a cohort of European individuals. For the European analysis, the authors used summary data from a recent meta-analysis of homocysteine genome-wide association studies (GWAS)12, (>44,000 European individuals) to generate an IV for circulating levels of homocysteine, of which they used to test the causal effect of homocysteine on blood pressure (BP) using summary data from the International Blood Pressure Consortium (IBCP) (>69,000 European individuals)13, in a two-sample MR approach. MR results showed that systolic blood pressure (SBP) decreased by 1.8mmHg (95% CI: -3.9, 0.4; P=0.11) in the Pelotas cohort but increased by 0.6mmHg (95% CI: -0.8, 1.9; P = 0.41) in the European population with each standard deviation (SD) increase in circulating log(homocysteine) levels. Similarly, diastolic blood pressure (DBP) increased by 0.1mmHg (95% CI: -1.5, 1.7; P=0.93) in the Pelotas cohort but increased by 1.1mmHg (95% CI: 0.2, 1.9; P=0.01) in the European population.
In another MR analysis using the same 1982 Pelotas Birth Cohort, Hartwig et al.14 found that lactase persistence (i.e. milk consumption) in adults was positively associated with BMI (effect estimate per 1dL/day increase in milk intake: 0.17kg/m2; 95% CI: 0.07, 0.27; P=0.001) and risk of obesity (OR per 1dL/day increase in milk intake: 1.09; 95% CI: 1.02, 1.17; P=0.015), contrary to observational estimates in the same cohort.Milk intake in Brazil (and potentially other similar Latin American countries) may predispose individuals to having a higher level of adiposity, a result that differs from inconsistent observational estimates derived from European populations.
These two examples alone underscore both the need for MR analyses to be conducted in LMICs, and the issues faced when dealing with genetic data. Firstly, large sample sizes are required for adequate statistical power in MR analyses due to the usually relatively small portion of variance explained in the risk factor by the genetic IV. The former described example, assessing the causal effect of circulating levels of homocysteine on BP, emphasises this particular point. Furthermore, a recent GWAS in over 340,000 individuals found that 66 genetic variants associated with BP in Europeans were also predictive of BP in 64,000 non-European samples; however, comparably large non-European sample sizes were necessitated to show this concordance between the direction of effect estimates and elucidate which genetic variants were associated with a specific ancestry group15. Therefore, the noticeable different sample size between the 1982 Pelotas Birth Cohort (N=3,701) and IBPC (N>69,000) in the aforementioned study, may partly explain the opposing direction of effect generated between the two populations. The relative cost of genotyping arrays and processing presents a risk of limited statistical power due to smaller sample sizes in MR analyses, particularly in LMICs, where appropriately sized samples may be relatively sparse.
Secondly, LMICs typically possess a multi-ethnic population structure (as seen in the Pelotas cohort in examples described above), showing a high level of genetic admixture and heterogeneity. The effect of risk factors on diseases that would typically affect one ancestral population (like Europeans, for example) might be irrelevant for an LMIC population as a result of this. In a recent study by Zanetti et al.16, global effect estimates for the association between SNPs and several common diseases, such as T2D, were typically in the same direction between different ancestral populations. However, the varying linkage disequilibrium structure (i.e. non-random association of genetic variants) between different ancestries largely influences the magnitude of effect between these populations. Ultimately, the difference in magnitude of the point estimates and likely heterogeneous population in Latin American countries underscores the extent to which MR analyses conducted in European populations may not represent the effects of the same exposures on outcomes in LMIC populations.
Whilst no studies have assessed the causal relevance of any exposure on T2D in LMICs using MR methodology, the described MR examples not only highlight their feasibility in LMICs, but also suggest that such studies will provide effect estimates more pertinent to LMICs. As more MR studies are published in LMICs, they could potentially refine population-specific IVs that take into account ancestral background, effect estimates and causal risk factors for relevant diseases such as T2D.
In order for future MR analyses in LMICs to be efficacious, there are certain properties, principals and limitations that should be taken into account, which have been outlined and discussed in detail previously6. Particular to LMIC settings, admixture and population heterogeneity due to multi-ethnic background can introduce genetic confounding and produce biased results. This can be addressed using methods of ancestral genomic control implemented in GWAS. Furthermore, selection bias and generalisability of findings (especially in populations where oversampling in low-socioeconomic status groups is likely) are of particular importance in practice. Finally, such studies will also rely on the availability of genetic data, high computational power and appropriate infrastructural facilities to store, maintain and analyse data required for MR analyses.
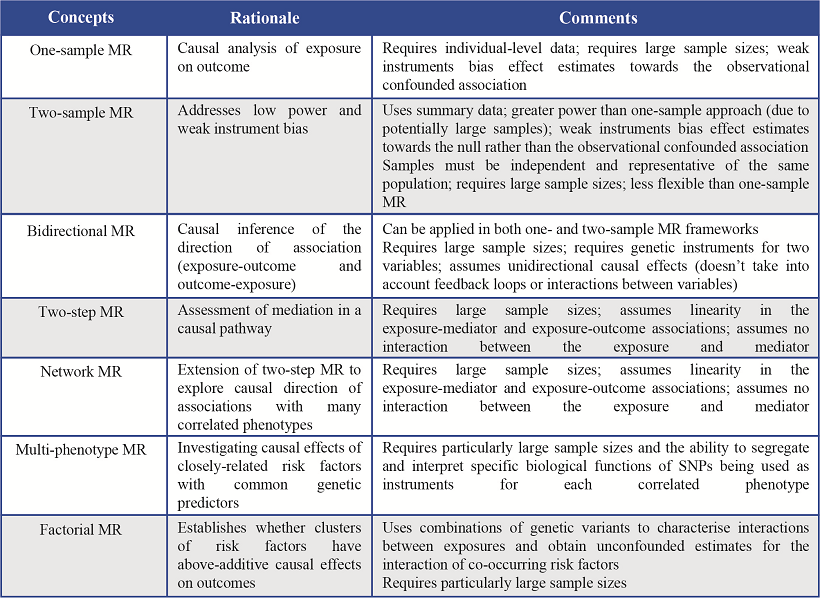
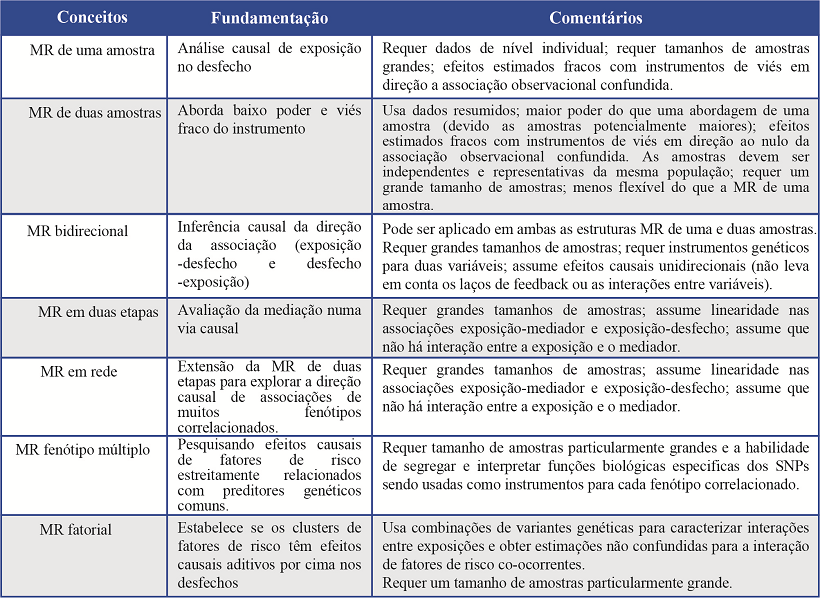
In the past decade or so, MR methodology has been increasingly applied to improve causal inference in a range of epidemiological contexts. Recent methodological developments building on the basic concept of MR, such as two-sample, two-step, network and multi-phenotype MR have made such complex analyses easily accessible for the research community, and provided more tools to dissect causal networks between traits with greater statistical power (Table 1)6,7.
Table 1. Methodological concepts, recent developments, strengths and limitations of Mendelian randomization analyses
Given the high prevalence of T2D in Latin America and LMICs, it is important to generate greater understanding of the potentially modifiable risk factors of T2D (along with other high-prevalence diseases and adverse health outcomes). With economic growth, availability of human tissue, increasing cost-effectiveness of genotyping arrays and recent developments in MR methodology, MR analyses are becoming ever-more practicable and could prove to be of fundamental importance when attempting to find outstanding risk factors for T2D that are particularly pertinent in LMICs.
La carga mundial de diabetes tipo 2 (DT2) está en aumento, parcialmente facilitada por un marcado incremento de la enfermedad en países de bajos y medianos ingresos (PBMI)1,2. Los PBMI no solo muestran una alta prevalencia de DT2 (8.7 %), pero han evidenciado un aumento más acelerado en esta prevalencia en los últimos 30 años comparados con los países de altos ingresos (PAI)1. Los factores de riesgo convencionales para DT2 en los PAI, como alto índice de masa corporal (IMC)3, bajos niveles de actividad física4 y malos comportamientos alimentarios5, no dan cuenta por completo del mayor aumento en la prevalencia de DT2 en los PBMI2. Por lo tanto, los factores de riesgo para DT2, específicamente dentro de un contexto de PBMI, deben ser determinados.
En la actualidad, se están utilizando análisis epidemiológicos observacionales para explicar la creciente prevalencia y determinar los factores de riesgo sobresalientes para DT2 en los PBMI, pero aun si están bien diseñados, dichos estudios son propensos a la confusión, causalidad inversa y múltiples fuentes de sesgo (por ejemplo, selección y medición)6. Como tal, estos diseños de estudio pueden potencialmente generar estimaciones poco fiables de causalidad entre un factor de riesgo y la enfermedad. Un enfoque exitoso y más robusto para superar estas limitaciones y mejorar la inferencia causal es la aleatorización Mendeliana (AM)6,7.
Brevemente, como resultado de los análisis de variable instrumental (VI) en econometría, La AM explota la primera y segunda leyes de herencia de Mendel (es decir, la distribución independiente y la segregación de alelos que conlleva a la distribución aleatoria de los genotipos en la población), permitiendo el uso de variantes genéticas para sustituir un rasgo clínicamente relevante (y usualmente modificable) (por ejemplo, IMC)6,7. Siempre y cuando se cumplan una serie de supuestos (Figura 1), estas VI genéticas pueden luego ser utilizadas para derivar el efecto causal sobre un rasgo en la enfermedad o resultado adverso para la salud, confiriendo múltiples ventajas sobre estimaciones de asociación derivadas de observaciones.
Figura 1. Gráfico acíclico dirigido de la teoría y supuestos clave de la aleatorización Mendeliana. Una variante genética (o variantes, G) se pueden utilizar como variables instrumentales para una exposición de interés (E) para evaluar la asociación causal entre E y el resultado de interés (O), dado que los siguientes tres supuestos se mantienen: (VI1) G debe ser ampliamente asociada con E; (VI2) G no debe ser asociada con ninguna variable de confusión medida o no medida (C); y (VI3) no debe haber asociación independiente entre G y O, dado E y C.
Análogas a los brazos de un ensayo clínico aleatorizado (ECA), las variantes genéticas utilizadas en la AM son ampliamente independientes de los factores de confusión debido a la naturaleza aleatoria de su asignación dentro de una población. Éstas tampoco son modificadas por el consiguiente desarrollo de la enfermedad o resultado de la salud (Figura 2) y, con el advenimiento de matrices de genotipificación más precisas, se reduce sustancialmente el error de medición. Por lo tanto, a nivel poblacional, la porción de varianza en el rasgo modificable explicado por las variantes genéticas utilizadas como una VI (contrario a la medición directa del rasgo) está libre de las limitaciones que de otro modo debilitaría la inferencia causal en estudios observacionales. La AM proporciona una estimación robusta y sin confusión de la asociación causal entre un rasgo y la enfermedad6,7.
Figura 2. Metodología de aleatorización Mendeliana comparada a ensayos clínicos aleatorios (ECAs). En un ECA, los individuos se asignan al azar a los brazos de tratamiento o de control (placebo), lo cual teóricamente asegura que los factores de confusión sean igualmente distribuidos entre los brazos del ensayo. Análogamente a esto, la asignación aleatoria y la distribución independiente de alelos en la concepción y la meiosis, respectivamente, asegura que los factores de confusión sean igualmente distribuidos entre los grupos de genotipos. Las diferencias en los individuos entre grupos de genotipos sólo se diferenciarán debido al grupo de genotipo; por consiguiente, podemos utilizar variantes genéticas como variables instrumentales para exposiciones de interés en los análisis de aleatorización Mendeliana.
Dentro del contexto de los PAI, la AM ha confirmado e identificado los factores de riesgo endógenos para DT2, incluyendo la obesidad, inflamación sistémica y lípidos en la sangre, y los factores de riesgo exógenos, incluyendo consumo de alcohol y lácteos8,9. Por ejemplo, el uso de la AM en una evaluación exhaustiva reciente de la asociación causal entre el IMC y la DT2 produjo estimaciones causales refinadas, que sugieren que el riesgo para DT2 aumentó aproximadamente 30% con cada unidad (kg/m2) en el IMC (odds ratio (OR): 1.26; intervalo de confianza (IC) del 95 %: 1.19; 1.32; P = 3,29 x 10-10)10. Sin embargo, estos hallazgos no pueden traducirse fiablemente a los PBMI. Hasta recientemente, el costo y la disponibilidad de grandes muestras de datos genéticos y fenotípicos restringieron el análisis de la AM a los PAI; una mayoría de los cuales residen dentro de poblaciones de ascendencia europea. Por tanto, aunque es probable que una mayor adiposidad puede, al menos en parte, explicar la creciente prevalencia de la diabetes en los PBMI, la estimación de la asociación no se ha probado y, actualmente, sólo existen unos pocos estudios de AM que evalúan el impacto de cualquier exposición modificable sobre riesgo de enfermedad especialmente en los PBMI.
Específicamente dentro del contexto de Latinoamérica, Borges et al.11 investigaron el efecto causal de la circulación de los niveles de homocisteína en la presión arterial en la Cohorte de Nacimientos de Pelotas de 1982 en Brasil utilizando la AM, luego compararon estos resultados a los de una cohorte de individuos europeos. Para el análisis europeo, los autores utilizaron datos resumidos de un meta-análisis reciente de estudios de asociación de todo el genoma (GWAS, por sus siglas en inglés)12, de homocisteína, (>44.000 individuos europeos) para generar una VI para los niveles circulantes de homocisteína, los cuales se utilizaron para comprobar el efecto causal de la homocisteína sobre la presión arterial (PA) utilizando datos resumidos del Consorcio Internacional sobre Presión Arterial (International Blood Pressure Consortium, IBCP) (>69.000 individuos europeos)13, en un enfoque de AM de dos muestras. Los resultados de AM demostraron que la presión sanguínea sistólica (PSS) disminuyó por 1.8 mmHg (IC 95 %: -3.9; 0.4; P = 0.11) en la cohorte de Pelotas, pero aumentó por 0.6 mmHg (IC 95 %: -0.8; 1.9; P = 0.41) en la población europea con cada desviación estándar (DE) aumenta en los niveles circulantes de log(homocisteína). Igualmente, la presión arterial diastólica (PAD) aumentó por 0.1 mmHg (IC 95 %: -1.5; 1.7; P = 0.93) en la cohorte de Pelotas, pero aumentó por 1,1 mmHg (IC 95 %: 0.2; 1.9; P = 0.01) en la población europea.
En otro análisis de AM utilizando la misma Cohorte de Nacimientos de Pelotas de 1982, Hartwig et al.14, encontró que la persistencia de la lactasa (es decir, consumo de leche) en adultos fue asociada de manera positiva con el IMC (efecto estimado por aumento de 1 dL/día en la ingesta de leche: 0.17 kg/m2; IC 95 %: 0.07; 0.27; P = 0.001) y riesgo de obesidad (OR por aumento de 1 dL/día en la ingesta de leche: 1.09; IC 95 %: 1.02; 1.17; P = 0.015), contrario a las estimaciones observacionales en la misma cohorte.La ingesta de leche en Brasil (y potencialmente otros países similares de Latinoamérica) puede predisponer a las personas a presentar un nivel más alto de adiposidad, un resultado que difiere de las estimaciones observacionales inconsistentes derivadas de las poblaciones europeas.
Estos dos ejemplos por sí solos subrayan la necesidad de que el análisis de AM se lleve a cabo en los PBMI, y los asuntos confrontados al tratar con datos genéticos. Primero, se requieren grandes tamaños de muestra para un poder estadístico adecuado en el análisis de AM debido a la, usualmente, relativamente pequeña porción de la varianza explicada en el factor de riesgo por la VI genética. El anterior ejemplo descrito, que evalúa el efecto causal de la circulación de niveles de homocisteína en la PA, destaca este punto particular. Además, un reciente GWAS en más de 340.000 personas encontró que 66 variantes genéticas asociadas con PA en los europeos también fueron predictivos de PA en 64.000 muestras no europeas; sin embargo, se necesitaron tamaños de muestra no europeos comparativamente grandes para mostrar esta concordancia entre la dirección de los efectos estimados y dilucidar cuáles variantes genéticas se asociaron con un grupo de ascendencia específico15. Por lo tanto, el tamaño de muestra notablemente diferente entre la Cohorte de Nacimientos de Pelotas de 1982 (N= 3.701) y el IBPC (N >69.000) en el estudio mencionado, puede explicar, en parte, la dirección opuesta de efecto generado entre las dos poblaciones. El costo relativo de las matrices de genotipado y procesamiento presenta un riesgo de poder estadístico limitado debido a tamaños de muestra más pequeños en el análisis de la AM, particularmente en los PBMI, donde las muestras de tamaño apropiado pueden ser relativamente escasas.
Segundo, los PBMI típicamente poseen una estructura poblacional multiétnica (como se vio en la Cohorte de Pelotas en los ejemplos anteriormente descritos), mostrando un alto nivel de mezcla genética y heterogeneidad. El efecto de los factores de riesgo en las enfermedades que típicamente afectarían una población ancestral (como europeos, por ejemplo) podría ser irrelevante para una población de un PBMI como resultado de esto. En un estudio reciente por Zanetti et al.16, las estimaciones de efectos globales para la asociación entre SNPs (polimorfismos de nucleótido único) y varias enfermedades comunes, como DT2, estaban típicamente en la misma dirección entre diferentes poblaciones ancestrales. Sin embargo, la estructura variable de desequilibrio de ligamiento (es decir, asociación no aleatoria de las variantes genéticas) entre diferentes ascendencias influye en gran medida la magnitud del efecto entre estas poblaciones. Por último, la diferencia en la magnitud de las estimaciones puntuales y probable heterogeneidad de la población en los países de Latinoamérica subraya el grado en que el análisis de la AM realizada en las poblaciones europeas puede no representar los efectos de las mismas exposiciones en los resultados en las poblaciones de los PBMI.
Aunque ningún estudio ha evaluado la relevancia causal de cualquier exposición en DT2 en los PBMI mediante el uso de la metodología de AM, los ejemplos de AM descritos no sólo resaltan su factibilidad en los PBMI, pero también sugieren que dichos estudios proporcionarán estimaciones de efecto más pertinentes a los PBMI. A medida que se publican más estudios sobre la AM en los PBMI, éstos podrían potencialmente refinar las VI específicas a la población que tienen en cuenta los antecedentes ancestrales, los efectos estimados y los factores de riesgo causales para enfermedades relevantes, como DT2.
Para que el análisis futuro de AM en los PBMI sea eficaz, hay ciertas propiedades, principios y limitaciones que se deben tener en cuenta, que ya han sido esbozadas y discutidas en detalle previamente6. Particular para los escenarios de los PBMI, la mezcla y la heterogeneidad de la población debido al origen multiétnico pueden introducir confusión genética y producir resultados sesgados. Esto se puede abordar utilizando métodos de control genómico ancestral implementados en GWAS. Además, el sesgo de selección y la generalización de los hallazgos (especialmente en poblaciones en las que es probable que exista un sobre-muestreo en grupos de bajo nivel socioeconómico) revisten especial importancia en la práctica. Finalmente, tales estudios también dependerán de la disponibilidad de datos genéticos, alto poder computacional e instalaciones de infraestructura apropiadas para almacenar, mantener y analizar los datos requeridos para el análisis de la AM.
Durante la última década, la metodología de AM ha sido ampliamente aplicada para mejorar la inferencia causal dentro de un rango de contextos epidemiológicos. Los recientes desarrollos metodológicos basados en el concepto básico de la AM, como AM de dos muestras, dos etapas, en red y AM multi-fenotipo han hecho tales análisis complejos fácilmente accesibles para la comunidad de investigación, y han proporcionado más herramientas para disecar redes causales entre rasgos con mayor poder estadístico (Tabla 1)6,7.
Tabla 1. Conceptos metodológicos, desarrollos recientes, fortalezas y limitaciones de los análisis de la aleatorización Mendeliana

Dada la alta prevalencia de DT2 en Latinoamérica y los PBMI, es importante generar mayor comprensión sobre los factores de riesgo potencialmente modificables para DT2 (junto con otras enfermedades de alta prevalencia y resultados adversos para la salud). Con el crecimiento económico, la disponibilidad de tejido humano, aumento de la costo-efectividad de las matrices de genotipificación y los recientes desarrollos en la metodología de la AM, los análisis de la AM se hacen cada vez más prácticos y pueden resultar de fundamental importancia al tratar de hallar factores de riesgo sobresalientes para DT2 que son particularmente pertinentes en los PBMI.
Texto em Português
A carga global da diabetes tipo 2 (T2D, por suas siglas em inglês) está aumentando, em parte facilitado por um aumento acentuado da doença em países de baixa e média renda (LMICs, por suas siglas em inglês)1,2. LMICs não só mostra uma alta prevalência da DT2 (8.7%), senão que ainda tem mostrado um incremento muito mais rápido na sua prevalência nos últimos 30 anos quando comparada com os países com renda alta (HICs, por suas siglas em inglês)1. Os fatores de risco convencionais para a T2D nos HICs, tais como o índice de massa corporal elevado (BMI, por suas siglas em inglês)3, baixos níveis de atividade física4, e comportamentos alimentares maus5, não são completamente responsáveis pelo grande incremento na prevalência do T2D nos LMICs2. Por tanto, os fatores de risco para a T2D especificamente dentro do âmbito dos LMIC precisam ser determinados.
Atualmente, análises epidemiológicas observacionais estão sendo usadas para explicar a prevalência crescente e determinar os fatores de risco notáveis para a T2D nos LMICs, porém ainda estando bem desenhados, esses estudos tendem a ter confundimento, causalidade reversível e múltiplas fontes de viés (p.ex. seleção e mensuração)6. Sendo assim, estes desenhos de estudo podem potencialmente gerar estimativos de causalidade pouco fiáveis entre um fator de risco e uma doença. Uma abordagem mais robusta para superar estas limitações e melhorar a inferência causal é a randomização Mendeliana (MR, por suas siglas em inglês)6,7 .
Sucintamente, resultando das analises de variáveis instrumentais (IV, por suas siglas em inglês) em econometria, a MR explora a primeira e a segunda lei de herança de Mendel (isto é, o sortimento e segregação independente dos alelos que leva à distribuição aleatória dos genótipos na população) favorecendo o uso das variantes genéticas para serem usadas como substitutos para uma caraterística clínica (p.ex. BMI)6,7, relevante (e geralmente modificável). Previstas um número de premissas chave que se cumprem (Figura 1), estas IVs genéticas podem ser usadas para obter o efeito causal em uma caraterística na doença ou desfecho adverso de saúde, conferindo múltiplas vantagens sobre os estimados de associação derivados das análises epidemiológicos observacionais.
Figura 1. Grafo acíclico dirigido (DAG, por suas siglas em inglês) da teoria e premissas da randomização Mendeliana. A variante genética (ou variantes, G) podem ser usadas como variáveis instrumentais para uma exposição de interesse (E) para avaliar a associação causal entre E e o desfecho do interesse (O) dado que as próximas três premissas contêm: (IV1) G deve estar fortemente associada com E; (IV2) G não deve estar associada com nenhuma variável de confusão mensurada ou não mensurada (C); e (IV3) não deve haver uma associação independente entre G e O, dadas E e C.
Análogo aos braços de um ensaio clínico randomizado (RCT, por suas siglas em inglês), as variantes genéticas usadas na MR são amplamente independentes de fatores de confusão devido à natureza aleatória da sua distribuição dentro da população. Elas também não são modificadas pelo desenvolvimento tardio da doença ou desfechos de saúde. (Figura 2) e, com a vinda de variedades de genótipos mais precisos, o erro na mensuração é grandemente reduzido. Consequentemente, ao nível da população, a porção da variância na característica modificável por variantes genéticas usadas como uma IV (diferentes da mensuração direta da caraterística) é livre de limitações as quais de outra forma debilitam a inferência causal nos estudos observacionais. A MR fornece uma robusta, estimativa não confundida da associação causal entre a característica e a doença6,7.
Figura 2. A metodologia da randomização Mendeliana comparada com ensaios clínicos randomizados (RCTs, por suas siglas em inglês). Em um RCT, os indivíduos são randomizados para ambos os braços, tratamento ou controle (placebo), os quais teoricamente garantem que os fatores de confusão sejam distribuídos igualmente entre os braços do ensaio. Similar a isto, a alocação aleatória e variedade independente de alelos na concepção e a meiose, respetivamente garantem que os fatores de confusão sejam distribuídos igualmente entre os grupos de genótipos. As diferenças nos indivíduos entre grupos de genótipos diferirão só devido ao grupo de genótipo; logo, podemos usar variantes genéticas como variáveis instrumentais para exposições de interesse nas análises de randomização Mendeliana.
No contexto dos HICs, a MR há confirmado e identificado fatores de risco endógenos para a T2D incluído a obesidade, inflamação sistêmica y lipídios no sangue, e fatores de risco exógenos incluído o álcool e o consumo de lácteos8,9. Por exemplo, o uso da MR em uma avaliação abrangente recente da associação causal entre o BMI e a T2D produziu estimativas causais aprimoradas as quais sugeriam que o risco da T2D se incrementou em aproximadamente 30% com cada unidade (kg/m2) no BMI (razão de chances (OR, por suas siglas em inglês): 1.26, intervalo de confiança de 95% (CI, por suas siglas em inglês): 1.19, 1.32; P=3.29x10-10)10. Contudo, estes achados não podem ser fielmente aplicadas para os LMICs. Até há pouco, o custo e disponibilidade de grandes amostras de dados genéticos e fenotípicos restringiram as análises MR para os HICs; a maioria deles residem dentro de populações com ascendência europeia. Portanto, embora é bem possível que uma adiposidade maior, pelo menos em parte, explique a prevalência crescente da diabetes nos LMICs, a associação estimada permanece não testada e atualmente só existem uns poucos estudos de MR que avaliem o impacto de qualquer exposição modificável no risco de doença, especialmente nos LMICs.
Especificamente dentro do contexto da América Latina, Borges et al.11 investigaram o efeito causal dos níveis de homocisteína na pressão do sangue na Coorte de Nascimento de Pelotas, Brasil em 1982 usando a MR, logo compararam esses resultados com aqueles de uma coorte Europeia de indivíduos. Para a análise europeia, os autores usaram dados resumidos de uma meta-análise recente de estudos de associação ampla de genomas (GWAS, por suas sigas em inglês)12, de homocisteína, ( >44,000 indivíduos da Europa) para gerar uma IV para os níveis de circulação de homocisteína, os quais eles usaram para testar o efeito causal da homocisteína na pressão arterial (BP, por suas siglas em inglês) usando informação resumida do Consórcio Internacional de Pressão Arterial (ICBP, por suas siglas em inglês)13, ( >69,000 indivíduos da Europa), em uma abordagem de MR de duas amostras. Os resultados da MR mostraram que a pressão arterial sistólica (SBP, por suas siglas em inglês) decresceu em 1.8mmHg (95% CI: -3.9, 0.4; P=0.11) na coorte de Pelotas, porém aumentou em 0,6mmHg (95% CI: -0.8, 1.9; P=0.41) na população Europeia com cada aumento no desvio padrão (SD, por suas siglas em inglês) nos níveis de circulação log(homocisteína). Do mesmo modo, a pressão arterial diastólica (DBP, por suas siglas em inglês) aumentou em 0.1mmHg (95% CI: -1.5, 1.7; P=0.93) na coorte de Pelotas, porém aumentou em 1.1mmHg (95% CI: 0.2, 1.9; P00.01) na população Europeia.
Em outra análise de MR usando a mesma coorte de nascimentos de Pelotas de 1982, Hartwig et al14, encontraram que a persistência de lactase (p.ex. no consumo de leite) em adultos estava associada positivamente com o BMI (efeito estimado por 1dL/ incremento diário na ingestão de leite: 0.17kg/m2; 95% CI: 0.07, 0.27; P=0.001) e o risco de obesidade (OR por 1dL/ incremento diário na ingestão de leite: 1.09; 95% CI: 1.02, 1.17; P=0.015), ao contrário das estimativas observacionais na mesma coorte. A ingestão de leite no Brasil (e potencialmente de outros países similares da América Latina) poderia predispor aos indivíduos a terem um maior nível de adiposidade, um resultado que diverge das estimativas observacionais inconsistentes derivadas da população Europeia.
Estes dois exemplos, por si destacam ambas as necessidades de que análises de MR sejam conduzidos nos LMICs, e os problemas enfrentados quando tratando com os dados genéticos. Primeiro, grandes tamanhos de amostras são requeridos para um adequado poder estatístico nas análises de MR, devido à relativa pequena porção de variância explicada no fator de risco pela IV genética. O exemplo descrito anteriormente, avaliando o efeito causal dos níveis de circulação de homocisteína na BP, enfatiza este ponto em particular. Além disso, um GWAS recente em mais de 340.000 indivíduos achou que 66 variantes genéticas associadas com a BP em população Europeia também eram preditivas da BP em 64.000 amostras não europeias; no entanto, grandes tamanhos de amostras não europeias que sejam comparáveis serão precisados para mostrar esta concordância entre a direção das estimativas de efeito e elucidar quais variantes genéticas estavam associadas com um grupo específico ancestral15. Consequentemente, o notoriamente diferente tamanho da amostra entre a Coorte de Nascimento de Pelotas em 1982 (N=3,701) e a IBPC (N>69,000) no estudo acima mencionado, poderia explicar parcialmente a direção oposta do efeito gerado entre as duas populações. O custo relativo das variedades de genotipagem e o processamento apresentam um risco de poder estatístico limitado devido aos tamanhos de amostras mais pequenos nas análises de MR, particularmente nos LMICs, onde apropriados tamanhos de amostras poderiam ser relativamente poucos.
Segundo, os LMICs comumente possuem uma estrutura de população multiétnica (como pode-se ver na coorte de Pelotas no exemplo acima descrito), a qual mostra um grande nível de mistura genética e heterogeneidade. O efeito de fatores de risco em doenças que poderiam tipicamente afetar uma população ancestral (como os da Europa, por exemplo) poderiam ser irrelevantes para uma população nos LMIC como resultado disso. Em um estudo recente feito por Zanetti et al16, o efeito nas estimativas globais para a associação entre SNPs y várias doenças comuns, tais como a T2D, foram geralmente na mesma direção entre diferentes populações ancestrais. Contudo, a estrutura de desequilíbrio da variedade de ligações (p.ex. a associação não aleatória de variantes genéticas) entre ascendências diferentes influencia amplamente a magnitude do efeito entre estas populações. Finalmente, a diferencia na magnitude das estimativas do ponto e a provável população heterogênea nos países da América Latina, destacam a extensão do porquê as análises MR realizadas em populações europeias poderiam não representar os efeitos da mesma exposição em desfechos nas populações LMIC.
Embora nenhum estudo tem medido a relevância causal de qualquer exposição à T2D nos LMICs usando a metodologia MR, os exemplos MR acima descritos não só realçam sua viabilidade nos LMICs, senão também sugerem que tais estudos fornecerão efeitos estimados mais pertinentes para os LMICs. Quanto mais estudos MR sejam publicados nos LMICs, eles potencialmente poderiam refinar as IVs de populações específicas as quais levam em conta o contexto ancestral, os efeitos estimados e os fatores de risco causal para doenças relevantes como a T2D.
Para que futuros análises MR nos LMICs sejam eficazes, há certas propriedades, fundamentos e limitações que devem ser levados em conta, os quais têm sido descritos e discutidos em detalhe anteriormente6. É particular dos cenários dos LMIC, a mistura e a heterogeneidade da população devido a que o background multiétnico pode introduzir confundimento genético e produzir resultados enviesados. Isto pode ser abordado usando métodos de controle genômico ancestral implementado em GWAS. Além disso, o viés de seleção e a generalização dos achados (especialmente em populações onde é provável a sobre-amostragem em grupos de baixo nível socioeconômico) são de particular importância na prática. Finalmente, tais estudos também se apoiarão na disponibilidade dos dados genéticos, o alto poder computacional e locais de infraestrutura apropriados para guardar, manter e analisar os dados requeridos pelas análises MR.
Ao longo da década passada, a metodologia MR há sido aplicada cada vez mais para melhorar a inferência causal em uma série de contextos epidemiológicos. Desenvolvimentos metodológicos recentes construídos sob conceito básico da MR, tais como duas amostras, duas etapas, network e fenótipos múltiplos MR tornaram tais análises complexas de fácil acesso para a comunidade de pesquisadores, e forneceram mais ferramentas para analisar networks causais entre características com maior poder estatístico (Tabela 1)6,7.
Tabela 1. Conceitos metodológicos, desenvolvimentos recentes, fortalezas e limitações das análises de randomização Mendeliana
Dada a prevalência da T2D na América Latina e nos LMICs, é importante gerar um entendimento maior dos potenciais fatores de riscos modificáveis da T2D (junto com outras doenças de alta prevalência e desfechos adversos de saúde). Com o crescimento económico, a disponibilidade de tecido humano, crescente custo-efetividade de variedades de genótipos e desenvolvimentos recentes na metodologia MR, as análises MR estão se voltando ainda mais praticáveis e poderiam provar ser de importância fundamental na hora de fazer tentativas para achar fatores de risco extraordinários para a T2D que sejam particularmente pertinentes nos LMICs.
Funding: This work was supported by CRUK [grant number C18281/A19169].
Conflict of interest: The authors declare no conflict of interest.
REFERENCES/REFERENCIAS/REFERÊNCIAS